User Guide
The following subcommands are available in gaftools
find_path - Extracting base sequencing from node path.
gfa2rgfa - Converting GFA to rGFA using W lines.
index - Indexing a GAF file for the view command.
order_gfa - Ordering GFA file using BO and NO tags.
phase - Adding phase information of the reads from a haplotag TSV.
realign - Realigning GAF alignments using wavefront alignment.
sort - Sorting GAF alignments using BO and NO tags of the corresponding graph.
stat - Basic statistics of the GAF file.
view - Viewing and subsetting the GAF file.
To get help for all the subcommands, please use:
gaftools <subcommand> --help
Links to related resources:
Some of the tags used in GAF files are derieved from minimap2 defined here
Requirement Summary by Subcommands
Subcommands |
rGFA Required? |
Ordering Required? |
|---|---|---|
find_path |
No |
No |
gfa2rgfa |
No |
No |
index |
Yes |
No |
order_gfa |
No |
No |
phase |
No |
No |
realign |
No |
No |
sort |
Yes |
Yes |
stat |
No |
No |
view |
Yes |
No |
gaftools find_path
This subcommand retrieves the base seqeunce of paths in the given GFA.
Usage
The find_path subcommand takes 2 obligatory inputs, a GFA file and either a node path -p, --path (e.g., ">s82312<s82313" (with the quotes))
or file path which has node paths each on a separate line with --paths_file.
It returns the sequence of the path(s) by default but using the --fasta flag, the sequences will be returned as a FASTA file.
If one of the paths does not exist, the process will terminate, the user can use -k, --keep-going to skip the paths that do not exist and continue with the next path if available.
Find the genomic sequence of a given connected GFA path.
positional arguments:
GFA GFA file (can be bgzip-compressed)
options:
-h, --help show this help message and exit
-p, --path PATH GFA node path to retrieve the sequence (e.g., ">s82312<s82313") with the quotes
--paths-file PATHS_FILE
File containing the paths to retrieve the sequences for, each path on new line
-k, --keep-going Keep going after instead of stopping when a path does not exist
-o, --output OUTPUT Output file. If omitted, use standard output.
-f, --fasta Flag to output the sequence as a FASTA file with the seqeunce named seq_<node path>
gaftools gfa2rgfa
This subcommand creates rGFA equivalent of GFA files using the W lines present in the GFA.
For creating the rGFA tags for all the reference nodes, the minimum requirements (as specified below) should be given. We assume that the pangenome graph is based on a base reference assembly (as is the case with minigraph-based graph constructions methods).
For further tagging the non-reference nodes coming from assemblies added to the reference assembly, the seqfile can be provided and corresponding W lines should be available in the GFA (refer to ‘Format of seqfile’).
If the seqfile is not provided, then the order in which walks are given in the GFA will be used to tag the non-reference nodes. Users can provide their own order of assemblies in the seqfile, and the coordinates will be generated accordingly.
Note: If the proper seqfile is not provided, the tagging will not show the coordinates intended by the graph creator since the order of assemblies might be different.
Any node not found in the W lines will be tagged with the SN tag as unknown, the SO tag as 0, and the SN tag as -1.
If the GFA already has the reference nodes tagged (as is the case of the GFA generated by minigraph-cactus), then the --reference-tagged flag can be provided and they will not be retagged.
Minimum Requirements
The GFA should have a reference path defined in the Header lines with
RS:Ztag or given by the user with--reference-name. There should also be W-lines for the reference path.The reference path described by the W line should be cycle-free and oriented in the forward direction (path of the format
>node_1>node_2....>node_N)
Note: From the Human Pangenome Reference Consortium, the graphs generated using minigraph-cactus follow these requirements. The PGGB graphs do not.
Format of seqfile
The seqfile asked by the subcommand has two columns separated by a tab: <assembly name> <tab> <path to the assembly>.
This file is part of the minigraph-cactus graph generation pipeline (in case the user is attempting to convert the GFA of that pipeline).
The assembly names should be of the form <sample name>.<assembly number> which should correspond to W lines starting with W <tab> <sample name> <tab> <assembly name> ...
In the case that the assembly is not based on a sample (as is the case of the chm13-minigraph-cactus graph which has a W line for GRCh38 added later and vice-versa for the grch38-minigraph-catus graph), the seqfile should contain a line for GRCh38 and GFA should have a corresponding W line of format W <tab> GRCh38 <tab> 0 ...
Users can provide their own order of assemblies and the coordinates will be generated accordingly. The reference path should be cycle-free.
Note: The code does not require the actual assemblies. So the assemblies provided in the seqfile do not need to be downloaded.
Usage
The gfa2rgfa subcommand takes 1 obligatory input, a GFA file. It creates a rGFA file with appropriate tags.
usage: gaftools gfa2rgfa [-h] [--reference-name REFERENCE NAME] [--override-reference] [--seqfile SEQFILE] [--output rGFA] GFA
Converting a GFA file to rGFA format using the W-lines and the acyclic reference path. (e.g., minigraph-based graphs)
positional arguments:
GFA GFA file (can be bgzip-compressed). This GFA should have a W-line corresponding to the reference genome or the reference nodes have to be tagged already.
options:
-h, --help show this help message and exit
--reference-name REFERENCE NAME
The name of the reference genome given in the W-line. If --reference-name is not specified, it looks for reference name in H lines.
--override-reference Flag to override any nodes tagged in the GFA as reference nodes.
--seqfile SEQFILE File containing the sequence in which assemblies were given. Provide the seqfile given as part of running minigraph-cactus (only the first column is required). It has the format: <assembly_name><tab><assembly_path>. If not provided, the order of assemblies will be taken from
the GFA file. User-defined order of assemblies can also be given. There should be W lines for each assembly in the GFA.
--output rGFA Output rGFA (bgzipped if the file ends with .gz). If omitted, use standard output.
gaftools index
This subcommand creates a index file with the extension .gvi which is used by the view command to subset alignments.
View Index Structure
The index is a reverse look-up table with the keys being nodes in the graph and the values being the location of the alignments which have the nodes.
The index is stored as a Python dictionary with the node IDs (as strings) as keys and a list of offset values (as integers) as values for the key.
This dictionary is written to memory using the pickle module.
{
"node_1": [offset_1, offset_2, ...],
"node_2": [offset_3, offset_4, ...],
...
}
Usage
The index subcommand takes 2 obligatory inputs, a GAF alignment file and its corresponding rGFA graph. It creates an viewing index file with the
extension .gvi which is required by the view command.
usage: gaftools index [-h] [-o OUTPUT] GAF rGFA
Index a GAF file for the view functionality.
This script creates an inverse look-up table where:
- key: the node information
- value: the offsets in the GAF file where the node is present
positional arguments:
GAF GAF file (can be bgzip-compressed)
rGFA rGFA file (can be bgzip-compressed)
options:
-h, --help show this help message and exit
-o, --output OUTPUT Output GAF View Index (GVI) file. Default: <GAF file>.gvi
gaftools order_gfa
This subcommand establishes an order to the graph based on the “bubbles” in the graph. Here, we define the bubbles as biconnected components, i.e. not the strict definition of a bubble found in other papers.
The graph input here has to be an rGFA, with SN and SO tags.
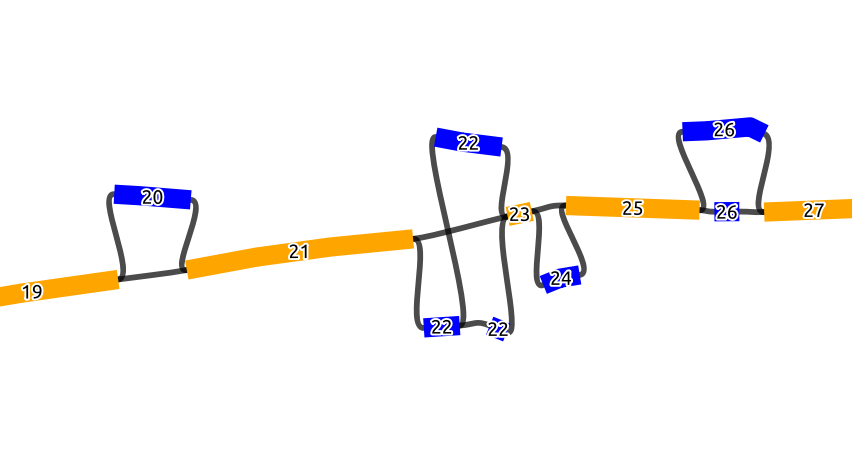
The basic idea here is that we first detect all biconnected components (bubbles), and collapse the detected bubbles into one node, which should result in a line graph made from scaffold nodes that belong to the reference and the collapsed bubbles inbetween. We then order this line graph in ascending order based on the coordinates in the SO tag. Each node in this ordered line graph gets an ascending BO tag from 1 to N (N being the number of nodes in the line graph). For the nodes that represent a collapsed bubbles, all the nodes in that bubble will get the same BO tag (Figure 1). As for the NO tag, the nodes in a bubble get an ascending number from 1 to M (M being the number of nodes in a bubble). However, the NO tag inside a bubble is assigned based on the node id order, i.e. in a lexicographic order of the node IDs. In the graph shown below, which is a part of a longer graph, when the bubbles collapsed, this will result in a line graph of 9 nodes.
Below we see a chain of 4 bubbles (biconnected components) and 5 scaffold nodes, where the nodes inside the bubbles are colored blue and the scaffold nodes are colored yellow. The numbers of the node is the BO tag, where it increases by 1 starting from the first scaffold node on the left (19 to 27), and we see that all the nodes in a bubble have the same BO tag
In this figure, we see the same graph but with the NO tags marked on the nodes. Scaffold nodes always have a NO tag of 0, and the nodes inside a bubble are marked with an increasing order of the NO tag.
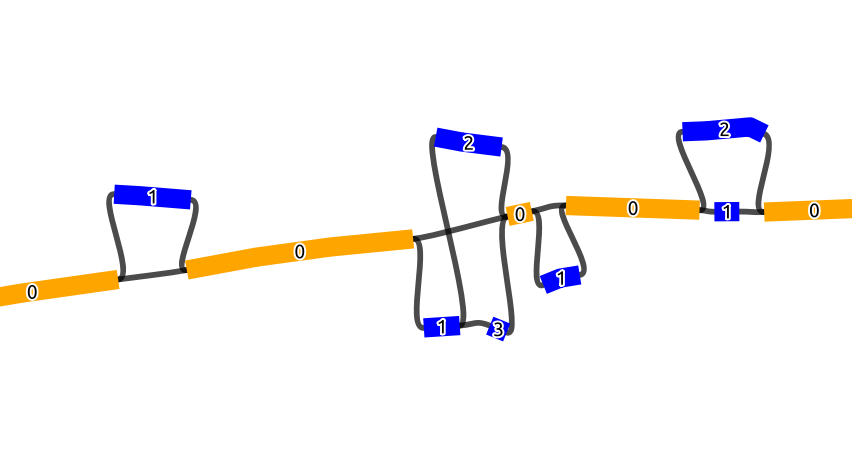
Note: The order_gfa subcommand does not change the name of the nodes and only adds the BO and NO tags as additional information.
Non-linear scaffold graphs:
In the cases where after collapsing the bi-connected component, the scaffold graph is not a line graph, the user can use
--ignore-branching to enforce an ordering where only the linear traversals in the scaffold graph where the articulation points
have an SN tag matching the reference will be ordered and branches that belong to other haplotypes will be ignored, i.e., give BO and NO tag of -1.
Usage
The order_gfa subcommand takes an rGFA as an obligatory input to order.
Optionally, the user can specify the order/subset of the chromosomes in the output GFA using the --chromosome-order option.
By default, the chromosomes are ordered in a lexicographic order of the component names.
With the --by-chrom flag, all the chromosomal graphs are output separately.
Users can also specify an output directory.
The outputs of order_gfa are separate rGFA graphs for each chromosome and a graph for all chromosomes both ordered by S lines first then L lines, and the S lines are ordered by
their BO tag then NO tag, also will output a CSV file with node colors similar to the figure above that works with Bandage.
usage: gaftools order_gfa [-h] [--chromosome-order CHROMOSOME_ORDER] [--without-sequence] [--outdir OUTDIR] [--by-chrom] [--ignore-branching] [--output-scaffold] GRAPH
Order bubbles in the GFA by adding BO and NO tags.
The BO (Bubble Order) tags order bubbles in the GFA.
The NO (Node Order) tags order the nodes in a bubble (in a lexicographic order).
positional arguments:
GRAPH Input rGFA file
options:
-h, --help show this help message and exit
--chromosome-order CHROMOSOME_ORDER
Order in which to arrange chromosomes in terms of BO sorting. By default, it is arranged in the lexicographic order of identified component names. Expecting comma-separated list. Example: 'chr1,chr2,chr3'
--without-sequence Strip sequences from the output graph (for less memory usage and easier visualization). Default = False
--outdir OUTDIR Output Directory to store all the GFA and CSV files. Default location is a "out" folder from the directory of execution.
--by-chrom Outputs each chromosome as a separate GFA, otherwise, all chromosomes in one GFA file
--ignore-branching Force the order even when branching paths occur in the scaffold graph. Alternative alleles will not be ordered
--output-scaffold Output the scaffold graph in GFA format. The scaffold graph is the graph created from collapsing all the biconnected components.
gaftools phase
This subcommands adds the phase information of the GAF reads from a haplotag TSV file generated using
whatshap haplotag.
Usage
The phase subcommand takes 2 obligatory inputs, a GAF alignment file and a haplotag TSV file generated from whatshap haplotag.
The TSV file has tags for each read labelled as H1, H2, or none for reads that has been determined to belong to the first haplotpye,
second haplotype or unknown haplotype. It also has the PS tag which is the phaseset that the read is in. Refer to WhatsHap documentation for
further details.
The phase command adds these tags to the GAF file so that downstream processes can utilize them.
usage: gaftools phase [-h] [-o OUTPUT] GAF TSV
Add phasing information to the GAF file from a haplotag TSV file.
The script uses the TSV file containing the haplotag information generated from WhatsHap's haplotag command.
The H1 and H2 labels for each read are then added to the reads in the GAF file.
positional arguments:
GAF GAF file (can be bgzip-compressed)
TSV WhatsHap haplotag TSV file. Refer to https://whatshap.readthedocs.io/en/latest/guide.html#whatshap-haplotag
options:
-h, --help show this help message and exit
-o, --output OUTPUT Output GAF file (bgzipped if the file ends with .gz). If omitted, output is directed to stdout.
gaftools realign
This subcommand realigns all the alignments in GAF back the rGFA it was originally aligned to using Wavefront Alignment. This fixes alignment issues found in GraphAligner where large indels are represented as a series of small indels in the CIGAR string.
Usage
The realign subcommand takes 3 obligatory input files, the GAF alignments, the rGFA graph that was used for the alignments,
and the reads that correspond to the alignments in the GAF file.
Due to the high memory consumption of pyWFA with longer alignments, gaftools limits the alignments to 60,000 base pairs in length
and the alignments that are longer will be outputted as is from the input file.
Moreover, realign can be sped up by using more cores. However, for longer alignments, memory can peak substantially, so users should be aware
that they need to maybe use a cluster with sufficient memory. For example, we tested alignments that were between 50,000 and 60,000 bp long, and when
gaftools was given 10 cores, the memory peaked to around 100 Gb at certain points, and with 1 core, it peaked at around 20 Gb. In case one of the subprocesses gets killed
by the system due to high memory consumption, the realignment run will be aborted.
usage: gaftools realign [-h] [-o OUTPUT] [-c CORES] GAF GFA READS
Realign a GAF file using wavefront alignment algorithm (WFA).
positional arguments:
GAF GAF file (can be bgzip-compressed)
GFA GFA file (can be bgzip-compressed)
READS Reads file in FASTA or FASTQ format (optionally gzipped)
options:
-h, --help show this help message and exit
-o, --output OUTPUT Output GAF file (bgzipped if the file ends with .gz). If omitted, use standard output.
-c, --cores CORES Number of cores to use for alignments.
gaftools sort
This subcommand sorts the alignments in the GAF file using the BO and NO tags generated by gaftools order_gfa. Hence this
subcommand requires initial processing of the rGFA with order_gfa.
Along with a sorted GAF file, the command also creates a sorting index based on the command flags.
Sort Index Structure
The current index is just pointers to the start and end position of alignments to chromosomes in the sorted GAF file.
The index is stored as a Python dictionary with the chromosome names (as strings) as keys and the start offset and end offset of the alignments in the GAF file as values.
This dictionary is written to memory using the pickle module.
{
"chr1": [start_offset_1, end_offset_1],
"chr2": [start_offset_2, end_offset_2],
...
}
Usage
The sort subcommand takes 2 obligatory input files, the GAF alignments, the rGFA graph that was used for the alignments.
The rGFA graph should have been processed by the order_gfa command and has the BO and NO tags.
The output GAF file is sorted based on the BO and NO tags of the rGFA graph and has additional tags added to each alignment. The
tags in the GAF are bo:i (which is the BO tag of the first node of the alignment), sn:Z
(which is the name of the reference chromosome the read aligned to), and iv:i (which is 1 is the alignment has an inversion).
By default, the GAF is outputted in stdout and without a sorting index. The flags --outgaf and --outind
can be used to provide path to the output GAF and its sorting index. If no --outind is given, the command automatically creates one
with the name of the GAF file provided and .gsi extension.
usage: gaftools sort [-h] [--outgaf OUTGAF] [--outind OUTIND] GAF GFA
Sort the GAF alignments using BO and NO tags of the corresponding graph.
positional arguments:
GAF GAF File (can be bgzip-compressed)
GFA GFA file with the sort keys (BO and NO tagged). This is done with gaftools order_gfa
options:
-h, --help show this help message and exit
--outgaf OUTGAF Output GAF (bgzipped if the file ends with .gz). If omitted, use standard output.
--outind OUTIND Output GAF Sorting Index file. When --outgaf is not given, no index is created. If it is given and --outind is not specified, it will have same file name with .gsi extension)
gaftools stat
This subcommand returns basic statistics of the GAF alignments like number of primary and secondary alignments, total aligned bases, average mapping quality, etc.
Usage
The stat subcommand takes 1 obligatory inputs, a GAF alignment file. It outputs statistics for the GAF file in the
stdout by default. The --cigar flag can be provided for more detailed statistics but requires more time.
usage: gaftools stat [-h] [-o OUTPUT] [--cigar] GAF
Calculate statistics of the given GAF file.
positional arguments:
GAF GAF file (can be bgzip-compressed)
options:
-h, --help show this help message and exit
-o, --output OUTPUT Output file. If omitted, use standard output.
--cigar Outputs cigar related statistics (requires more time)
gaftools view
This subcommand helps view the GAF alignments, convert formatting from stable to unstable and vice-versa, and subsetting the files based on nodes or regions given by the user.
Note: The subcommand currently does not support the ds tag that has been introduced in minigraph v0.21. The subcommand ignores that tag and outputs the rest of the fields.
Usage
The view subcommand takes 1 obligatory input, the GAF alignment file. But for full functionaility, it requires
the rGFA file which was used for the alignment and the index file created with gaftools index.
By only providing the GAF file, the view command can output the entire file.
- Additional files required for certain functionality:
Subsetting alignments based on region (given with
--regionor-r) or node (given with--nodeor-n) requires the viewing index generated bygaftools index.Converting coordinate system with
--formator-frequires an GFA file.
- Modes of selecting alignments (when giving multiple nodes or regions):
Union selects alignments that align to at least one of the nodes or regions.
Intersection selects alignments that align to all of the nodes or regions.
usage: gaftools view [-h] [-g GFA] [-o OUTPUT] [-i INDEX] [-n NODE] [-r REGION] [-f FORMAT] [-m MODE] GAF
View, subset or convert a GAF file (GAF file should be indexed first, using gaftools index, for subsetting).
The view command allows subsetting the GAF file based on node IDs or regions available.
positional arguments:
GAF GAF file (can be bgzip-compressed)
options:
-h, --help show this help message and exit
-g, --gfa GFA GFA file (can be bgzip-compressed). Required when converting from one coordinate system to another.
-o, --output OUTPUT Output GAF (bgzipped if the file ends with .gz). If omitted, use standard output.
-i, --index INDEX Path to GAF Viewing Index file. Required when subsetting alignments based on region or nodes. This index is created using gaftools index. If path is not provided, it is assumed to be in the same directory as GAF file with the same name and .gvi extension (default location of
the index script)
-n, --node NODE Nodes to search. Multiple can be provided (Eg. gaftools view .... -n s1 -n s2 -n s3 .....).
-r, --region REGION Regions to search. Multiple can be provided (Eg. gaftools view .... -r chr1:10-20 -r chr1:50-60 .....).
-f, --format FORMAT Format of output path (unstable | stable)
-m, --mode MODE Mode of selecting alignment with multiple regions/nodes provided ("I" | "U"). Choose "I" for finding alignments with all provided regions/nodes (intersection) and "U" for finding alignments with at least one of the provided regions/nodes (union). (default: U)